

Movement disorders, dopamine, cerebral folate deficiency, and thiamine

Prefer to listen to the podcast? Subscribe and listen free:
Dopamine is a fascinating and complex molecule influencing gross motor control, motivation, learning, and mood. It plays a role in many psychiatric and neurological conditions, which I have seen firsthand.
In this article, I will discuss how folate and thiamine influence dopamine and why I believe these two nutrients are behind many of the symptoms we’ve experienced over the last couple of years.
I shared my thoughts in this article with our daughter's neurometabolic physician, who agreed with most of what I believed to be occurring. I say most because I pushed back on a couple of things he said, and after a few exchanges, it appeared as if he didn’t think I was wrong; however, he wasn’t entirely sure if I was right either. I felt grateful even to have the opportunity to discuss at that caliber with someone who’s been in the game for so long!
I’ll share what we came up with and the rationale behind everything at the end of the article!
Dopamine and movement
Aside from the autoimmunity, the first sign that something else was wrong with our daughter was the delay in hitting her gross motor skills milestones. She technically met the milestones; however, they would occur towards the tail end of normal. Even after she began to walk, I noticed she had an unsteady gait and frequently fell (sometimes, this would happen from a standstill). The gait abnormalities would fluctuate, making it difficult for the many doctors we saw to appreciate what I would see at home.
Shortly after her first birthday, we noticed the first signs of dystonia (abnormal eye and jaw movements). Initially, I didn’t realize this was related to dopamine, and we were even reassured that this was normal in many children.
As a parent, I felt differently.
She was also a toe walker from the start, and it became more pronounced as she grew. I would later find out this, too, was related to dopamine. Eventually, she reached a point where we saw abnormal movements of her legs, trunk, arms, and hands. The symptoms fluctuated and were often worse in the evening; dopamine levels are highest in the morning and slowly decline throughout the day. Even so, she had signs of dopamine deficiency and excess, with the former being the predominant feature:
Dopamine deficiency
The following are signs and symptoms we’ve noticed signifying a dopamine deficiency:
Oculogyric crisis: paroxysmal, conjugate, tonic, usually upwards, deviation of the eyes. This would occur for seconds to minutes, depending on the day, and is almost always accompanied by mandibular dystonia (lateral movement of her jaw). I also noticed that Chloe would have an emotional/aggressive outburst seemingly out of the blue, followed by dystonic reactions 15-30 minutes later.
Paroxysmal tonic upgaze is another possibility. However, as stated in my previous post, I believe this is nothing more than semantics. Many neurologists believe this to be a variation of an oculogyric crisis that is perhaps more brief in nature (lasting seconds in some cases). Still, it is primarily considered to be secondary to a dopamine deficiency.
Toe walking and toe curling: dystonia localized to the ankle/foot resulting from a dopamine deficiency in the striatum.
Cerebellar ataxia: deficiency can lead to motor function/coordination loss. We’ve also experienced a few episodes of horizontal nystagmus lasting for several minutes.
Absence seizures/staring spells: When she was closer to one year of age, we noticed she would have multiple episodes daily (average of 10). In the middle of an activity, she would stare off into the distance and remain unresponsive for about 20-30 seconds. It would self-resolve, and she would continue doing the activity as if nothing had happened. However, these staring spells might have resulted from decreased dopamine levels. It has been demonstrated that dopamine agonists suppress absence seizures.
It’s worth noting that a folate deficiency can also result in absence seizures, as it can impair the synthesis of GABA, resulting in a glutamate/GABA imbalance. This imbalance in the thalamocortical circuits of the brain is thought to be part of the pathophysiology of absence seizures.
Acute dystonic reaction: About 4-6 weeks back, she had an abrupt onset of pain in her throat (not associated with eating). She would grab her throat, say she has pain, and then it would quickly resolve after 15 seconds. I’m not exactly sure what this was, but it happened with enough frequency for me to notice; this no longer occurs.
Dopamine excess
We’ve noticed chorea as well. This is a state of excessive, spontaneous movements that are irregularly timed, non-repetitive, randomly distributed, and abrupt. The timing of these movements can be further described as athetosis (slow) and choreoathetosis (intermediate), but they aren’t all that helpful in guiding management. As noted above, this affects her legs, trunk, arms, and hands.
Chorea can also result from an acetylcholine deficiency, which she is also at risk for due to her genetics. When a folate deficiency occurs, choline is converted to betaine and used in the methylation cycle to compensate for the lack of folate. Because of this, a choline deficiency can occur, leading to a decreased production of acetylcholine.
As this is the only dopamine excess symptom, it’s also possible that her chorea resulted from a decreased production of acetylcholine.
Diurnal fluctuation
It’s important to note that her signs of dopamine deficiency would improve with sleep (this is a classic pattern). This is commonly seen because dopamine production increases with each REM sleep cycle.
So, the next question we have to ask is why she has a dopamine deficiency. To answer that, let’s review how dopamine is created.
Dopamine biosynthesis
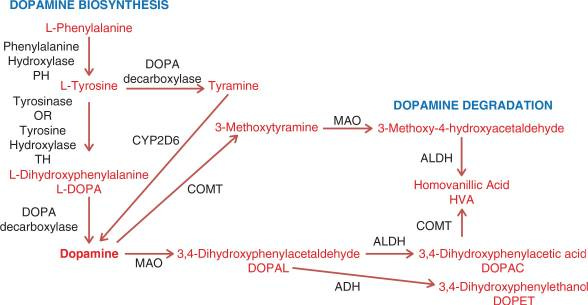
Everything starts with the essential amino acid phenylalanine. The image above shows the steps needed to convert this amino acid into the neurotransmitter dopamine.
The conversion of phenylalanine to tyrosine is catalyzed by the phenylalanine hydroxylase (PAH) enzyme, using tetrahydrobiopterin (BH4), iron, and NADH as cofactors. Suppose this conversion can’t occur (due to a deficiency in the PAH enzyme or any of the cofactors). In that case, a tyrosine deficiency will ensue, ultimately leading to a deficiency in dopamine.
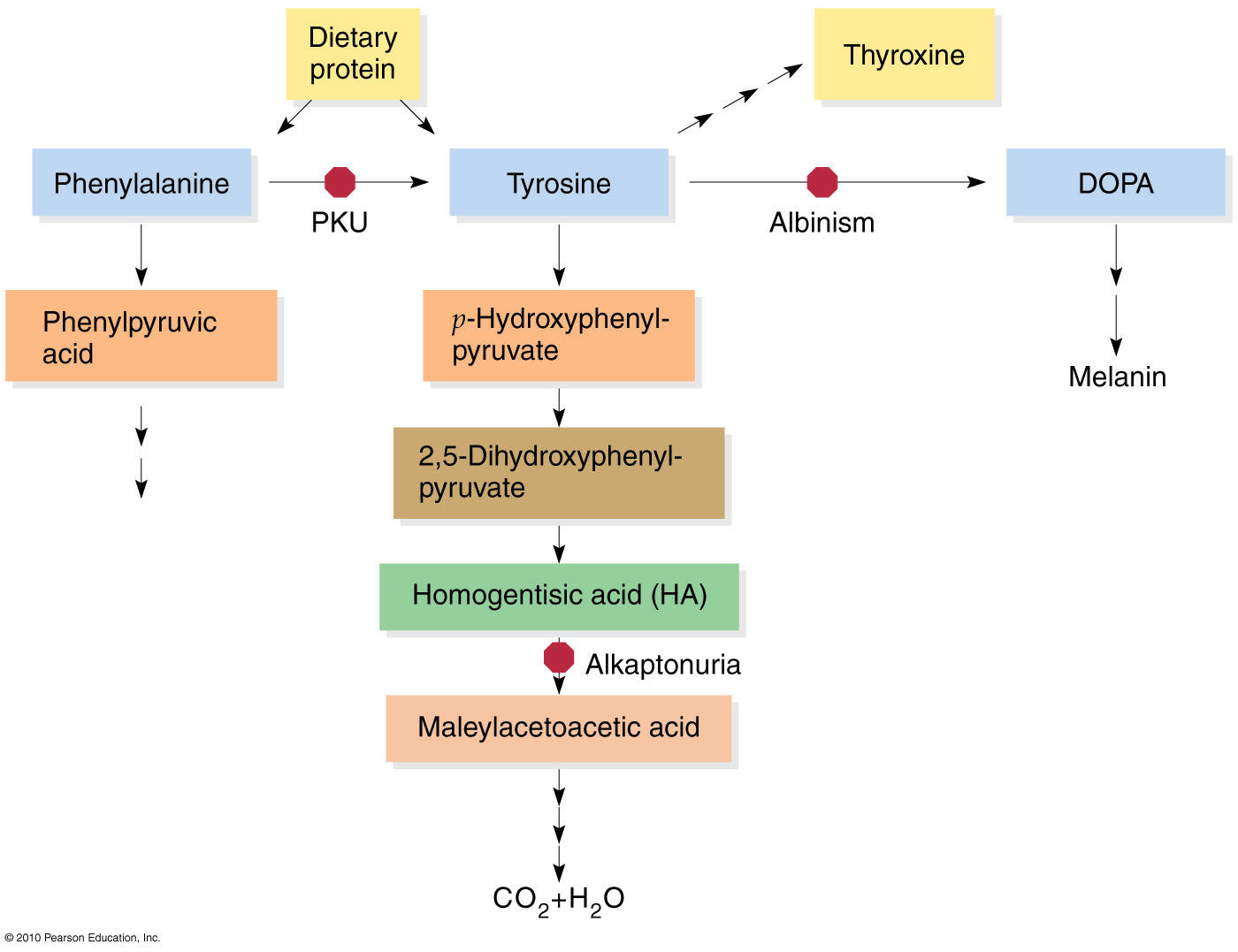
Remember that this block's severity can be mild, moderate, or complete. In other words, if you have a complete block, then zero phenylalanine is converted into tyrosine and results in hyperphenylalaninemia (as seen in classic phenylketonuria); this forces phenylalanine to be transaminated into phenylpyruvate, phenylacetate, and/or phenyllactate (as depicted below):

Side note: You’ll notice that tyrosine can be converted to tyramine and subsequently to dopamine via the CYP2D6 enzyme. However, it’s unclear how much this pathway contributes to the overall biosynthesis of dopamine.
After tyrosine is synthesized, it then needs to be converted to levodopa via the tyrosine hydroxylase enzyme, along with the same cofactors as the previous step. Suppose this conversion can’t occur (due to a deficiency in the tyrosine hydroxylase enzyme or any of the cofactors). In that case, a levodopa deficiency will ensue, ultimately leading to a deficiency in dopamine.
If a complete block is present, then zero tyrosine is converted into levodopa and results in hypertyrosinemia; this forces tyrosine to create 4OH-phenylpyruvate or go down another pathway to create thyroxine for thyroid hormone synthesis:
Levodopa is then converted to dopamine, which is catalyzed by DOPA decarboxylase (DDC). This reaction uses pyridoxal 5' phosphate (PLP) and iron as cofactors. A deficiency in any cofactor or the DDC enzyme results in elevated levodopa and decreased dopamine levels.
Phenylalanine to Tyrosine
So, we first want to look at phenylalanine and its conversion to tyrosine. Below, you can see that our daughter has normal phenylalanine but decreased tyrosine. A normal phenylalanine/tyrosine ratio typically ranges from 0.5 to 1.5. My daughter has a ratio of approximately 1.86:

An increased ratio suggests a problem with the conversion of phenylalanine to tyrosine. A phenylpyruvate elevation (as seen on her urine organic acid tests) also supports this assumption.

You’ll notice her phenylalanine is normal, but remember that the blocks don’t have to be complete, which means a partial conversion is still occurring.
Although her phenylalanine is normal, a partial block can still increase phenylpyruvate levels (see image above). It’s also unclear what her phenylalanine levels are in the fed state; perhaps postprandial phenylalanine levels exceed the postprandial reference range.
In any event, I believe we can rule out a phenylalanine hydroxylase deficiency (problem with the enzyme) because she had a normal ratio in June 2023, along with a negative newborn screening and whole genome sequencing:

So, now we know that she has a problem converting phenylalanine to tyrosine, and this is most likely due to a problem with a cofactor:
Tetrahydrobiopterin (BH4)
Iron
NADH
Tetrahydrobiopterin (BH4) and Folate
Our daughter is homozygous for the MTHFR C677T mutation, which means she has a decreased capacity for synthesizing 5-methyltetrahydrofolate (5-MTHF). Because 5-MTHF helps recycle BH2 back to BH4, a deficiency would result in low BH4 levels.
She is also homozygous for the CBS C699T mutation, which results in elevated ammonia levels. The body will use the limited amount of BH4 to clear ammonia (this a priority), leaving less than ideal amounts for serotonin and dopamine.
Even though our daughter was receiving 3,950 mcg of folate, the following support that she was still deficient:
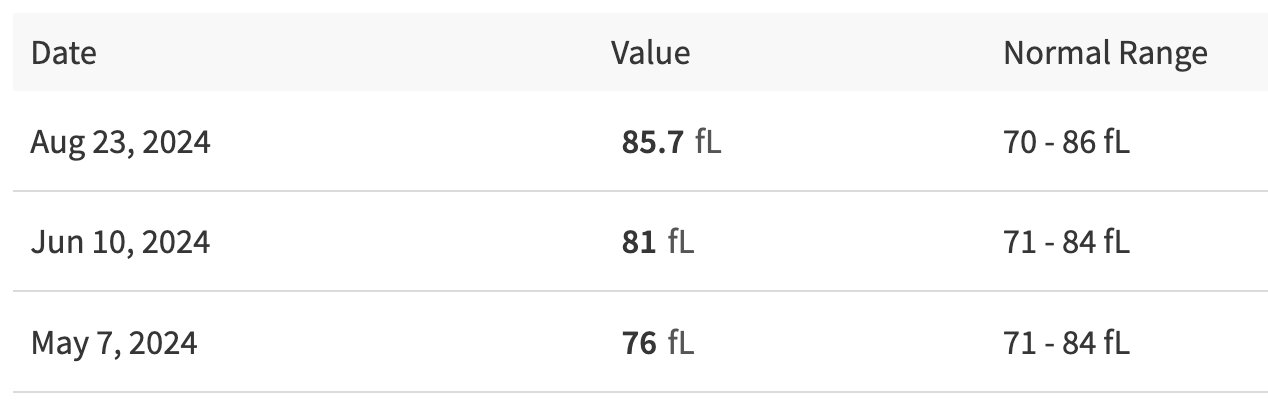
Her MCV has steadily increased:
Elevated sarcosine levels: Folate is required to convert sarcosine to glycine. Therefore, a folate deficiency will prevent this, resulting in an elevated sarcosine level.

At this point, we know she has a problem converting phenylalanine to tyrosine, most likely from a tetrahydrobiopterin deficiency (BH4). I believe she has low BH4 levels because she has low folate levels.
Many might ask if it’s possible to have a folate deficiency with normal folate levels.
Absolutely, it’s possible!
Cerebral Folate Deficiency
We have three ways of routinely assessing for a folate deficiency:
Serum folate levels
RBC folate levels
Homocysteine
Our daughter has normal RBC folate and homocysteine levels:
RBC folate: 641 ng/mL (reference Range: >280 ng/mL)
Homocysteine: 3.0 umol/L (reference Range: <10.4 umol/L)
Although there is no lower limit of normal, I believe the optimal range is much closer to 5-7 umol/L (a lower level could be secondary to a lack of resources, i.e., methionine)
We must remember that serum levels don’t always equate to CSF levels. Cerebral folate deficiency is characterized by decreased CSF 5-MTHF levels, which can occur in normal or elevated RBC and serum folate.
Patients can present with any developmental or neuropsychiatric disorder, often in the form of psychomotor regression (losing social, mental, or motor skills); this has been implicated as a potential cause/contributing factor in autism.
Cerebral folate deficiency occurs because there is a disruption in the folate receptor-alpha (FRα) transporter, which is responsible for transporting folate across the choroid plexus into the CSF. This can be a result of:
Genetic defects affecting transporter function (FOLR1)
Autoantibodies bind to the receptor and prevent transport
Mitochondrial dysfunction as this is dependent on ATP
We have a mitochondrial mutation and have had problems with autoantibodies (at one point, she met the criteria for lupus). A deficiency can also occur due to increased cerebral folate requirements (any time inflammation or infection is present) or decreased availability (as in the case of her MTHFR mutation).
Two months ago, our daughter was completely asymptomatic and was on 6,250 mcg of folate (roughly 50% from folinic acid and 50% from 5-MTHF).
Unfortunately, we discontinued her supplements in July because of the taste. I spent over a month reformulating her entire supplement cocktail, and I’ve had to titrate back up to her previous doses slowly. Currently, we are on 5,550 mcg of folate (850 mcg from folinic and 4,700 mcg from 5-MTHF).
I have considered increasing her folinic acid to her previous dose but hesitated because of her MTHFR mutation. Folinic acid still requires conversion to 5-MTHF via the MTHFR enzyme. Because she is homozygous for this mutation, she has a 70% reduction in this enzyme activity.
I’m concerned that adding folinic acid would result in a large amount of unmetabolized folate, which could negatively affect the balance between glutamate and GABA. This is because folate is comprised of pterin, p-aminobenzoic acid (PABA), and glutamate:

In theory, this could have a positive effect, as it could increase tetrahydrobiopterin (BH4) levels, as folate is nothing more than a conjugated pterin, and BH4 is an unconjugated pterin. Also, 5-MTHF eventually gets recycled back to tetrahydrofolate (THF) anyway. But I have still struggled.
Before proceeding, I wanted to hear what our mitochondrial specialist had to say. So, I presented this article to our neurometabolic doctor, who agreed with everything I wrote. He is also concerned about a cerebral folate deficiency. However, he feels our daughter will do better with an increased folinic acid dose. In fact, he wants to add 5,000 mcg of folinic acid (split twice daily) to her current regimen for a total dose of 10,000 mcg.
He believes this will be transported into the CSF much more efficiently than 5-MTHF; even with her MTHFR mutation, the mega dose of folinic acid will allow enough to be converted and raise levels of 5-MTHF in the CSF. He isn’t too concerned with what would occur with the unmetabolized portion.
His rationale has to do with the various folate transporters that exist:
Folate Receptor Alpha (FRα): This is the primary transporter responsible for delivering 5-MTHF into the CSF.
Proton-coupled folate Transporter (PCFT): This plays a secondary role in folate transport, particularly in acidic environments. It can transport 5-MTHF; however, it has a higher affinity for folinic acid as it can be transported more efficiently (the methyl group on 5-MTHF affects transport).
Reduced Folate Carrier (RFC): This low-affinity transporter plays a minor role in transporting folates into the CSF. It can transport 5-MTHF. However, this, too, has a higher affinity for folinic.
Because cerebral folate deficiency is considered to be a defect with folate receptor-alpha, our neurometabolic specialist believes that folinic acid provides better transport into the CSF via PCFT, even if it has a decreased conversion to 5-MTHF once inside the CSF.
Even so, he ordered an EEG to rule out any seizure activity. So, we plan to do that in the next couple of weeks.
Thiamine and Dopamine
Aside from folate, I have seen remarkable improvements in mega-dosing with thiamine. Thiamine has been shown to increase dopamine levels through various mechanisms (it is beneficial in Parkinson's disease).
Thiamine pyrophosphate (TPP) is a cofactor for pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase in the citric acid cycle. These pathways are critical for producing NADH (cofactor to synthesize tyrosine and l-dopa) and ATP (dopamine synthesis is a high-energy activity).
It is also a cofactor for the enzyme transketolase used in the pentose phosphate pathway to generate NADPH. This helps protect neuronal cells against oxidation and increases dopamine synthesis.
Thiamine has also been shown to directly enhance the release of dopamine (possibly mediated through calcium-dependent channels).
Due to reformulating her supplement regimen, we removed the thiamine HCL and thiamine tetrahydrofurfuryl disulfide (TTFD). We also decreased the total thiamine dose from 1,300 mg to 485 mg. Just like the folate, we have started to titrate back up according to the following schedule:
8/24: 615 mg (285 mg thiamine mononitrate and 330 mg benfotiamine)
8/26: 765 mg (285 mg thiamine mononitrate and 480 mg benfotiamine)
8/29: 915 mg (285 mg thiamine mononitrate and 630 mg benfotiamine)
8/31: 1,065 mg (285 mg thiamine mononitrate and 780 mg benfotiamine)
Only so much active and passive absorption can occur with thiamine HCL, thiamine mononitrate, and benfotiamine. For this reason, I was also using tetrahydrofurfuryl disulfide (TTFD). Unfortunately, it tastes awful, lol! So, I’m holding off on this until she’s old enough to swallow a pill. Aside from the absorption, it has other properties that make it incredibly beneficial in neurological diseases.
Our neurometabolic doctor is skeptical regarding the absorption of any oral formulation and believes systemic therapy (IM or IV) is necessary to achieve optimal levels. But, he first wants to check a serum thiamine level. It should be noted that a free thiamine level isn’t the most sensitive test, but it’s easy and doesn’t require a specialized laboratory. It has been proposed that a more sensitive marker for thiamine deficiency is an erythrocyte transketolase activity and thiamine pyrophosphate effect. But we’ll start with a free thiamine level first.
Improvement in labs and symptoms
Starting September 1st, the emotional outbursts, chorea, oculogyric crisis, mandibular dystonia, ataxia, throat pain, and staring spells were entirely resolved.
Starting September 4th, she stopped waking up with dried blood in her nostrils and started intermittently walking heel-to-toe, although these moments were very brief. Our neurometabolic doc isn’t too concerned with the toe walking, as he says she’s only three. Still, the overall pattern of a dopamine imbalance leads me to think otherwise (similar to the dystonic eye movements).
After one week of adjusting her thiamine and folate dosages, I am so happy to share that, for the first time, we have a completely normal urine organic acid test. I should also mention that I’ve adjusted various other supplements based on her previous results (l-carnitine, choline, phosphatidylserine, and taurine, to name a few), which have also contributed to this beautiful picture courtesy of Quest:



Biochemistry for the win! Until next time!